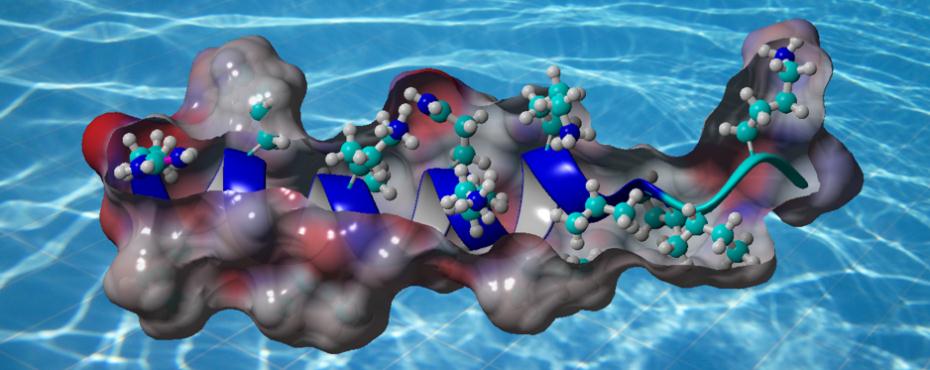
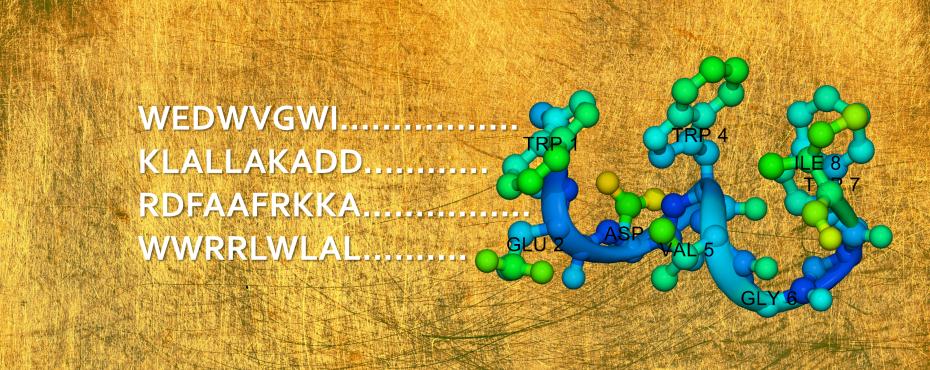
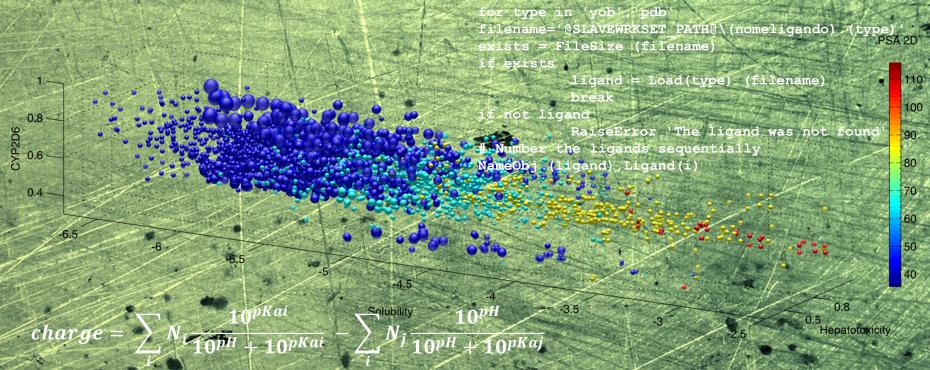
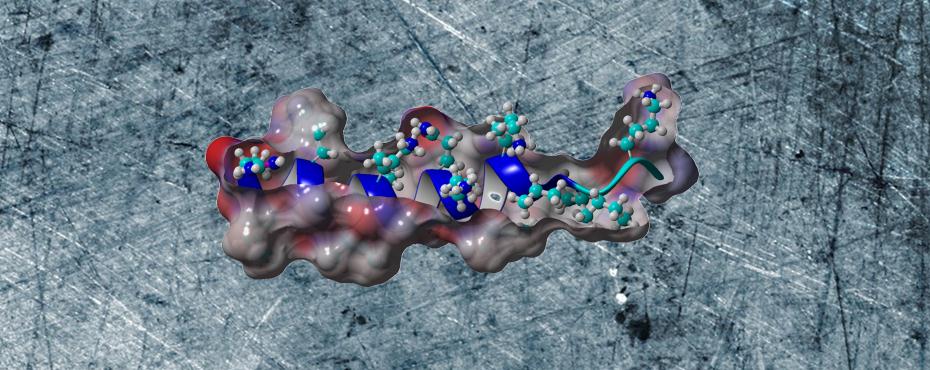
YADAMP
Home
About
Search Database
Theory
Stats
Literature
Contact
Previous
Next